Research
Our laboratory is interested in understanding the molecular and cellular mechanisms underlying Parkinson’s disease (PD). Pathogenic mutations in the LRRK2 gene are a common genetic cause of PD and induce increased LRRK2 kinase activity. Increased LRRK2 activity is also triggered by other PD-causing mutations, lysosomal membrane damage, and exposure to PD-causing environmental toxins. These findings indicate that elevated LRRK2 kinase activity is a common pathogenic driver of different forms of PD, but the intracellular pathways that are altered by hyperactive LRRK2 and mediate its pathogenic effects remain elusive.
Over the past years, we have made important contributions to our understanding of LRRK2-related PD. We were amongst the first to show that pathogenic mutations cause an increase in the LRRK2 kinase activity, suggesting that kinase inhibitors hold promise as disease-modifying compounds. We discovered LRRK2-mediated alterations in lysosomal functioning and autophagic membrane trafficking, now widely accepted as one of the disease-relevant phenotypes associated with LRRK2-related PD. We demonstrated the importance of calcium in mediating lysosomal dyshomeostasis due to pathogenic LRRK2, and we described a role for iron, which is known to accumulate in the brains of LRRK2-PD patients as compared to healthy controls.
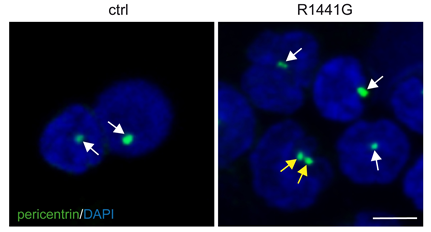
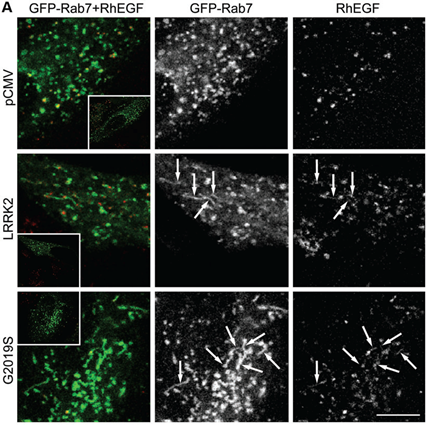
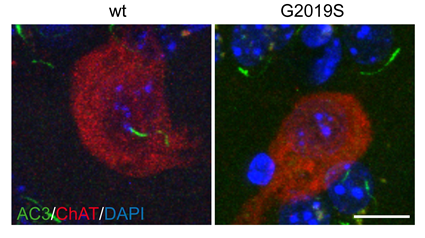
A subset of Rab proteins which are master regulators of intracellular membrane trafficking have been identified as LRRK2 kinase substrates. This led us to investigate how Rab protein phosphorylation impacts on membrane trafficking events. We found deficits in endosomal trafficking due to LRRK2-mediated Rab protein phosphorylation, which causes defects in the proper recycling of various receptors and transporters. We for the first time described centriolar alterations due to pathogenic LRRK2, including in peripheral cells derived from LRRK2-PD patients, and we are investigating whether this cellular readout can serve to stratify PD patients who may benefit from LRRK2 kinase inhibitor-related therapeutics. The centriolar deficits are due to the accumulation of phosphorylated Rab substrates, and such accumulation is also responsible for impaired ciliogenesis (a centrosome-related event) mediated by LRRK2, which comprises a disease-relevant phenotype in the intact rodent and human brain.
Our laboratory is investigating the underlying mechanism(s) and consequences of these cellular alterations using molecular, biochemical and high-resolution cell biological approaches in a variety of disease-relevant cell types, patient-derived cells and tissues, and transgenic mouse models. Our goal is to understand the molecular pathogenesis of PD, so as to aid in the development of novel therapies to treat this devastating disorder.