Cr51 Release Assay To Detect Cytotoxic Activity of CAR-T and CAR-NK cells
Author: Yan Yang and Dongfang Liu
Date: 02/06/2019
Reagents
Reagents |
Location |
Catalog number |
Note |
LumaPlatesTM |
C509 |
6006633 (PerkinElmer) |
|
Cr-51 (sodium chromate), 1mCi |
C509 |
102653-222 (VWR) |
5 mCi/ml |
TUMOR cell lines |
K562 cell line |
|
|
CAR-T/CAR-NK cells |
CD19-CAR |
|
|
96-well plates, V-shape |
C509 |
3894 (Corning) |
|
Triton-X |
|
|
5% |
Important Notes for Radio safety: For you and people around you
- Separate radioactive waste very carefully from regular waste.
- Monitor your work area after each use of this or any other radioactive isotope.
- Record usage of 51Cr every time.
- When the vial reaches half its total volume, it is a good idea to verify the actual amount of 51Cr in the tube -- sometimes there is less 51Cr in the vial because somebody forgot to record when they removed some.
Essential Background Knowledge
Chromium-51 (51Cr) release assays are commonly used for the precise and accurate quantification of cytotoxicity of NK cell and CTLs (Whiteside 2001, Sierich and Eiermann 2013), particularly in the study of tumor and viral cytolysis, as well as CAR T/NK cytotoxicity(Xiong, Chen et al. 2018). The assay is used to determine the number of lymphocytes produced in response to infection or drug treatment.
A brief overview of the assay principle is illustrated below. Target cells are labeled with 51Cr, the label is then released from the target cells by cytolysis. The label can be isolated by centrifuging the samples and collecting the supernatants. Supernatants from centrifugation can be dried on a LumaPlate™ and counted in a MicroBeta2 counter.
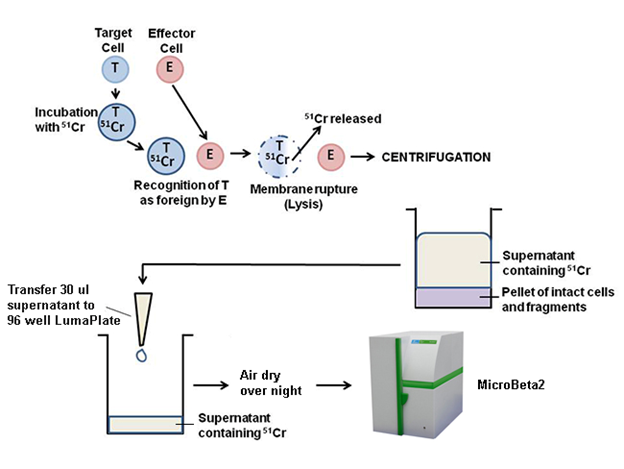
Experiment Design for Common 96-well Plate:
Terms |
Definition |
Effector cells |
NK, CAR-NK, CAR-T, or control cells (such as PBMC) |
Target cells |
TUMOR cell lines, here we use K562 cell line |
E/T ratio |
depending on potency of effector cells, sensitivity of target cells to lysis, and number of effector and target cells available. We usually start with 20:1 of E/T ratio. |
Spontaneous Release |
Target cells without Effector cells. Incubate Target cells with an equal volume of media |
Maximum Release |
Incubate Target cells with media containing 2.5% Triton X-100 |
CAR |
Chimeric antigen receptor |

Procedure
The main procedures including:
- Part 1: Radioactive labelling of target cells with 51Cr (Step 1).
- Part 2: Preparation of the effector cells (Step 2).
- Part 3: Load target cells into the assay plate (Step 3).
- Part 4: Transfer supernatant to LumaPlate (Step 4).
- Part 5: Measure the LumaPlate on the MicroBeta2 (Step 5).
- Part 6: Analysis of results (step 6).
Part 1: Radioactive labelling of target cells with 51Cr
- Calculate the number of target cells needed for the killing assay. You will need 1×104 target cells for 1 well of a 96-well plate. Consider adding 20% more cells to account for volume loss during pipetting.
Please use this plate to calculate how many cells you need:

- Harvest the required number of cells into 15 ml conical tube
- Spin cells down for 5 min at 350 × g.
- Wash the cells in 5 ml R-10
- Discard the supernatant and resuspend the cells in the remaining medium (about 200 µL).
- Add 50 µCi 51Cr (10 uL, activity 5 mCi/ml) per target, then resuspend cell with the same pipet tip you used to add the chromium. Pipets containing chromium should be discarded in the empty media/PBS bottle used for chromium pipet tip waste.
- Incubate cells at 37 ℃ in a 5% CO2 incubator for 2 hours. When we label K562 target cells for NK killing assay OVER Night, I incubate it over night because you will get the best result. For adherent cell line, such as SK-HEP1 and K562 TUMOR cell lines, we cannot incubate the adherent cells for overnight, because the cells may adhere to the tube or die from the non-adhesion. Therefore, 2-hour incubation time at 37 incubator with Cr51 is sufficient
Note:
- Take the necessary precaution and protection when manipulating radioactive material (e.g., lead shields, rapid manipulation).
- Do not vortex the cells, to avoid spontaneous release of 51Cr.
- The average radioactive activity measured in a maximum lysis control should not go below 900 counts per minute (cpm). If necessary, prolong the incubation time and/or increase the amount of 51Cr.
- For suspend-culture cell lines, such as K562 and 221 cells, please use retronectin to coat the 96-well plate (transparent flat bottom) over night. Before adding NK cells, please centrifuge the plate (Cr51-labeled 221 cells and K562 cells) 300 rpm for 5 min at room temperature.
Preparation of the effector cells (during the incubation of 51Cr labelling)
- Calculate the number of effector cells needed for the test.
- Count the effector cells with trypan blue and adjust to a density of 1 ×106 cells/ml.
- Take out 96-well rounded bottom plate and write all the necessary information on it. This will help you put the correct cells into the correct wells. Make sure to put markings on the plate itself as well as the lid to avoid confusion with the lids from multiple plates.
- Transfer 200 μl of each effector cells to triplicate wells in the first row (row A) of the 96-well plate.
- In the next five rows (rows B through F), pipet triplicates of 100 μl R-10. Transfer triplicates of 100 μl of each effector cells from row A to the 100 μl R-10 in row B. Resuspend thoroughly with a 100-μl multichannel pipettor and make a serial 1:2 dilution in the descending rows. Remove 100 μl and remain 100 μl medium for wells in row F after the dilution.
- Add 100 μl RPMI-10 medium to wells in row G (spontaneous release controls) and 100 μl 5% Triton X-100 to wells in row H (maximal release controls).
Load target cells into the assay plate
- At the end of 51Cr labeling time, wash the targets three times with 12 ml cold R10 at 350 × g for 5 min in a cold centrifuge.
- Discard the supernatant with the required safety precautions.
- After the third wash, resuspend the cells at a concentration of 1 × 105 cells/ml in RT RPMI/10% FBS.
- Add 100 µl target cells (1 × 104 cells) to each well using a multi-channel pipette.
- When the plate is filled, incubate it for 4 h at 37 °C in an incubator with 5% CO2.
Transfer supernatant to LumaPlate
- At the end of the 4-hour incubation, spin down the cells at 350 × g for 5 min. Then we need a centrifuge with 96-well plate capability.
- Transfer 30 µl of supernatant from each well to a LumaPlate using a multichannel pipette.
- Carefully note the order of transferred samples!
- Be sure that you do not touch the cell pellet. To avoid this, you may spin the plate(s) 5min at 350 × g, room temperature. However, even without centrifuging, the cells normallystick to the bottoms of the wells.
- Aspirate all liquid waste into the chromium vacuum flask and the dispose of the plates as chromium dry waste.
- Let the LumaPlate air dry overnight at room temperature. Why do we need to let the LumaPlate air dry?
- Measure the LumaPlate the next morning on the MicroBeta2.
Measure the LumaPlate on the MicroBeta2.
- Switch on MicroBeta2 with the power switch at the back of the counter.
- When the built-in computer is ready, and shows the Windows desktop,
- Start up the MicroBeta2 Windows Workstation program by clicking the desktop icon.
- Create a new protocol for yourself.
- Select the original protocol, “_Cr-51 Lumaplate”, to copy from the list.
- Click the Copy button. It will be given the name Copy of xxx where xxx is the name of the protocol copied. A dialog appears to allow you to edit parameters.
- Don’t forget your protocol ID. This number will be used on the barcode labels for cassette.
- When you have made the changes you want, save this protocol under its new name by clicking OK.
- Attach barcode labels for cassette and sample identification to the appropriate area of the ID support plate. If the amount of samples in an assay is such that more than one cassette is required, you only need to barcode the first cassette.
PROT (Protocol number)
This is normally a simple number in the range 0-99 (or STOP code, see below). The type of protocol it refers to depends on what is specified in the FUNC field.
- Fix a STOP code to the last cassette.
- Open the door on the front of the counter.
- Load the cassettes into the rack with the sample plate upwards and the cassette ID labels towards you.
- Place the cassette you want to count first on the bottom shelf, numbered 1, the cassette you want to count second on the next to the bottom shelf, numbered 2, and the remaining cassettes in the desired count order on shelves 3 to 16. The first cassette should be barcoded with appropriate PROT number. Don’t forget to put the cassette with STOP code on the top shelf.
Empty shelves between cassettes are allowed, and the first cassette you want to read does not have to be in position 1 as long as there are no other cassettes below it.
- After loading, close the door.
- When cassettes are barcoded, you can use the counter’s automatic start feature.
Click the Start button in the main window. If the system displays a message asking you to confirm the start of counting, click OK.
- After counting is complete, new results appear at the end of the Results list on the right of the Protocol group dialog.
An alternative way to access results is to click the Results icon on the main workstation toolbar. This opens the Results dialog which contains a list of all the results file names.
- To examine a result, select the desired result row and click Open in either the Protocol group dialog or the Results dialog.
- You can also print your results from the program used to open the generated output files.
Calculating the Results of the Chromium Release Assay
Once you have the readings, you need to perform a simple calculation to determine the cytotoxicity of your cells.
6.1 Average your triplicates for spontaneous release and maximum release. Then take those averages and plug them into this formula:
Spontaneous Release: Target cells without Effector cells. Incubate Target cells with an equal volume of media.
Maximum Release: Incubate Target cells with media containing 2.5% Triton X-100.
6.2 Load your data into the pre-programed Excel file for calculation.
6.2 Export the excel data into Prism for better graph.
Important Note:
It is very important to have effector and target cells in very good conditions. Target cells in bad condition (dying or dead) cannot be efficiently labelled with chromium, which leads to results that are difficult to interpret.
Assay Optimization
Experimental conditions such as the amount of radioactivity used to label target cells, the length of the target cell labeling incubation, E:T ratios, and E:T incubation times will vary based on cell types used and should be optimized for your particular assay.
Gamma vs. Beta Counting Efficiency
Traditionally, 51Cr is considered a gamma emitter. However, since 51Cr decays by electron capture it can be quantified by detection of the gamma ray in a gamma counter or by the detection of the more abundant Auger electrons and low energy x-rays in a liquid scintillation counting system. This is reflected in the counting efficiency in each method. In a gamma counter, counting efficiency is unlikely to be in excess of 7%. In an instrument such as the MicroBeta®, 51Cr counting efficiency is 26%.
References
Sierich, H. and T. Eiermann (2013). "Comparing individual NK cell activity in vitro." Curr Protoc Immunol Chapter 14: Unit 14 32.
Natural killer (NK) cells play an important role in the innate immune system by eliminating infected and mutated cells. Their cytotoxic capacities vary markedly among individuals. The cytotoxic activity can be measured in peripheral blood mononuclear cells (PBMCs) using the NK cell-specific target cell line K562. In this chapter, we present a protocol for the standardization and normalization of cell preparation and NK cell cytotoxicity measurement in a (51)Cr-release assay. By following these protocols, it is possible to compare the NK cell activity of numerous-if necessary selected-individuals in vitro.
Whiteside, T. L. (2001). "Measurement of cytotoxic activity of NK/LAK cells." Curr Protoc Immunol Chapter 7: Unit 7 18.
In this unit, methodology is presented for measuring the capacity of human natural killer (NK) or lymphokine-activated killer (LAK) cells to lyse tumor cell targets. Cytotoxicity of these effector cells is evaluated in a short-term (51)Cr-release assay using NK-sensitive tumor cells as the targets for NK cells or NK-resistant tumor cells as the targets for LAK cells. In the basic protocol, a generic (51)Cr-release assay is described in which PBMC, purified NK cells or interleukin 2 (IL-2)-activated lymphocyte populations (LAK cells) are utilized as effector cells. Support protocols describe preparation of nonadherent tumor cells, cells obtained from malignant effusions, trypsinized tumor cells from adherent monolayer culture, or freshly isolated tumor cells from surgical specimens. All of these cell types can serve as the (51)Cr-labeled target cells in the basic protocol. The procedure for labeling target cells from any of these sources with (51)Cr is also provided.
Xiong, W., et al. (2018). "Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells." Mol Ther 26(4): 963-975.
Chimeric antigen receptor (CAR)-modified T cell therapy has the potential to improve the overall survival of patients with malignancies by enhancing the effectiveness of CAR T cells. Precisely predicting the effectiveness of various CAR T cells represents one of today's key unsolved problems in immunotherapy. Here, we predict the effectiveness of CAR-modified cells by evaluating the quality of the CAR-mediated immunological synapse (IS) by quantitation of F-actin, clustering of tumor antigen, polarization of lytic granules (LGs), and distribution of key signaling molecules within the IS. Long-term killing capability, but not secretion of conventional cytokines or standard 4-hr cytotoxicity, correlates positively with the quality of the IS in two different CAR T cells that share identical antigen specificity. Xenograft model data confirm that the quality of the IS in vitro correlates positively with performance of CAR-modified immune cells in vivo. Therefore, we propose that the quality of the IS predicts the effectiveness of CAR-modified immune cells, which provides a novel strategy to guide CAR therapy.